Benchmarking scientific AI assistants with sequence-to-activity models
Since ChatGPT’s debut, a small but fast-growing industry of scientific AI assistant chatbots has emerged. Recently, Google released TxGemma, a suite of tools built on their Gemma models and fine-tuned with Therapeutics Data Commons. Others are trying to build companies around selling scientific AI assistant software.
The hard part is figuring out how to measure their performance. Last year, FutureHouse released the first dedicated benchmark, consisting of 2,457 questions and answers. I’ve been curious about a different angle for evaluating these models. Can we use models that predict variant effects from genome or protein sequence to evaluate the scientific AI assistants’ ability to understand literature?
Using one machine learning model to evaluate another is an example of model-guided evaluation. The key advantage to this approach is that it does not require manual curation of answers. Suppose we ask two assistants for two lists – disease‑causing missense variants and benign ones. A predictor such as AlphaMissense can then score those variants, giving us an external yard‑stick for how well each assistant separated signal from noise. While the variant effect predictor will not have perfect performance itself, we can assume the performance is constant across all LLMs tested1
As a proof-of-concept for this approach, I tested whether models are able to identify variants that cause splicing changes. I chose to focus on splicing variants because there are multiple high performing models, but the same approach could easily be applied to other classes of variants (e.g. missense, promoter, UTR).
Prompting for splicing variants
I focused on 16 AI assistants that have a web interface, and used the following two-sentence prompt:
Please provide me with a list of 10 human variants that cause splicing changes and 10 variants that do NOT alter splicing. I would like the genomic coordinates (hg38) of each variant, and please structure them in a data frame with columns: Chr, Position, Ref, Alt, Gene
To the extent possible, I avoided giving the models additional information and asked them to use their best judgement if they asked for clarifications. 11 of 16 models accepted the task and provided variant coordinates. The remaining four models provided general methodologies for how to find such variants, but were not willing to provide variant coordinates.
| Developer | Model | Accepted task |
|---|---|---|
| Anthropic | Claude 3.7 | ✅ |
| Anthropic | Claude 3.7 extended thinking | ✅ |
| Anthropic | Claude 3.7 web search | ✅ |
| Deep Origin | Balto | ✅ |
| Future House | Crow | ❌ |
| Microsoft | Gemini Deep Research | ✅ |
| OpenAI | GPT-4 | ❌ |
| OpenAI | GPT-4o | ✅ |
| OpenAI | Deep Research | ✅ |
| OpenAI | GPT-o1 | ✅ |
| OpenAI | GPT o3 | ✅ |
| OpenAI | GPT o4-mini | ✅ |
| Perplexity AI | Perplexity Pro | ❌ |
| Perplexity AI | Perplexity Deep Research | ✅ |
| Potato | Potato | ❌ |
| you.com | you.com | ❌ |
Are the model-provided variants real?
Before I could generate splicing predictions for the variants, I had to verify that they actually correspond to real genomic variants. I ran two checks:
- Does the listed reference base match the hg38 reference at the coordinate?
- Does the coordinate overlap the listed gene?
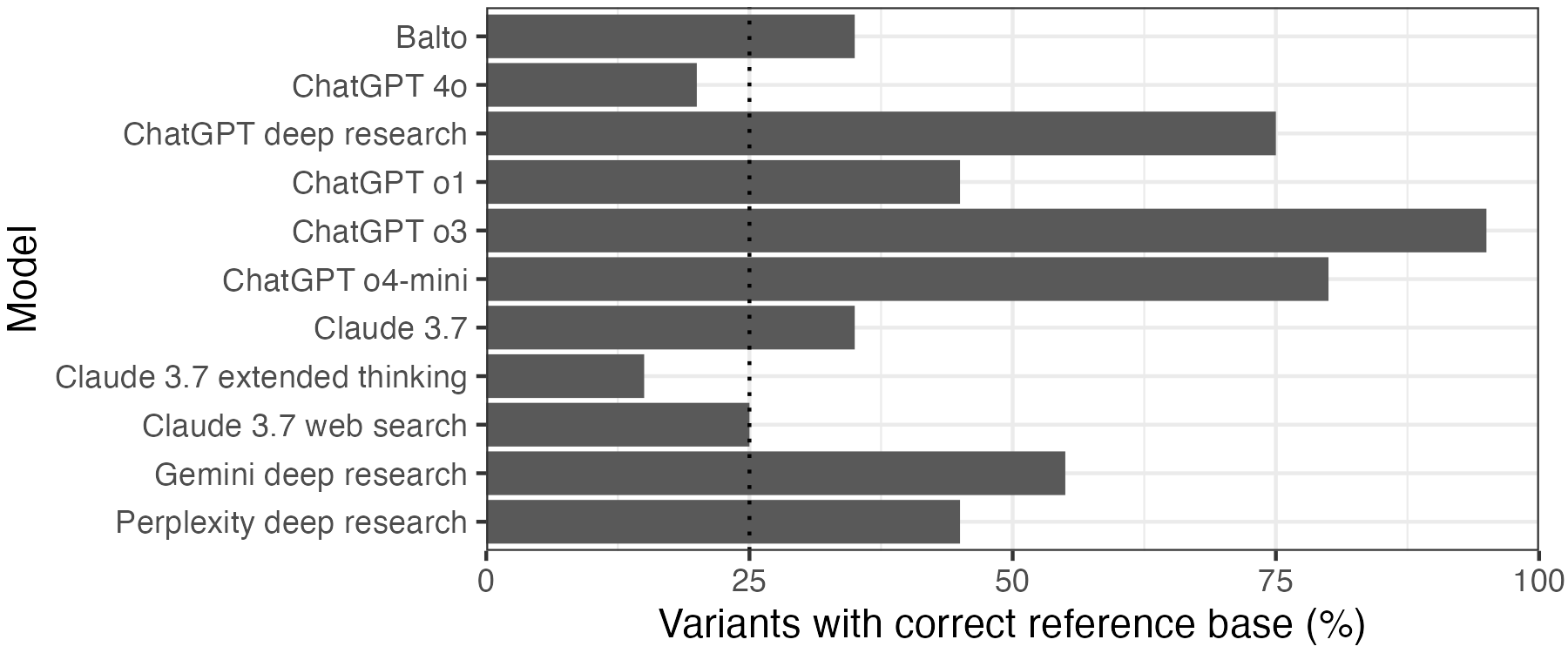
Here is where the wheels began to wobble. 4/11 models that provided coordinates did no better than random chance at identifying the reference base. This included ChatGPT 4o and Claude 3.7. By contrast, the reasoning models ChatGPT o3 and o4-mini, as well as ChatGPT’s deep research functionality, got the reference base for over 75% of the variants correct.
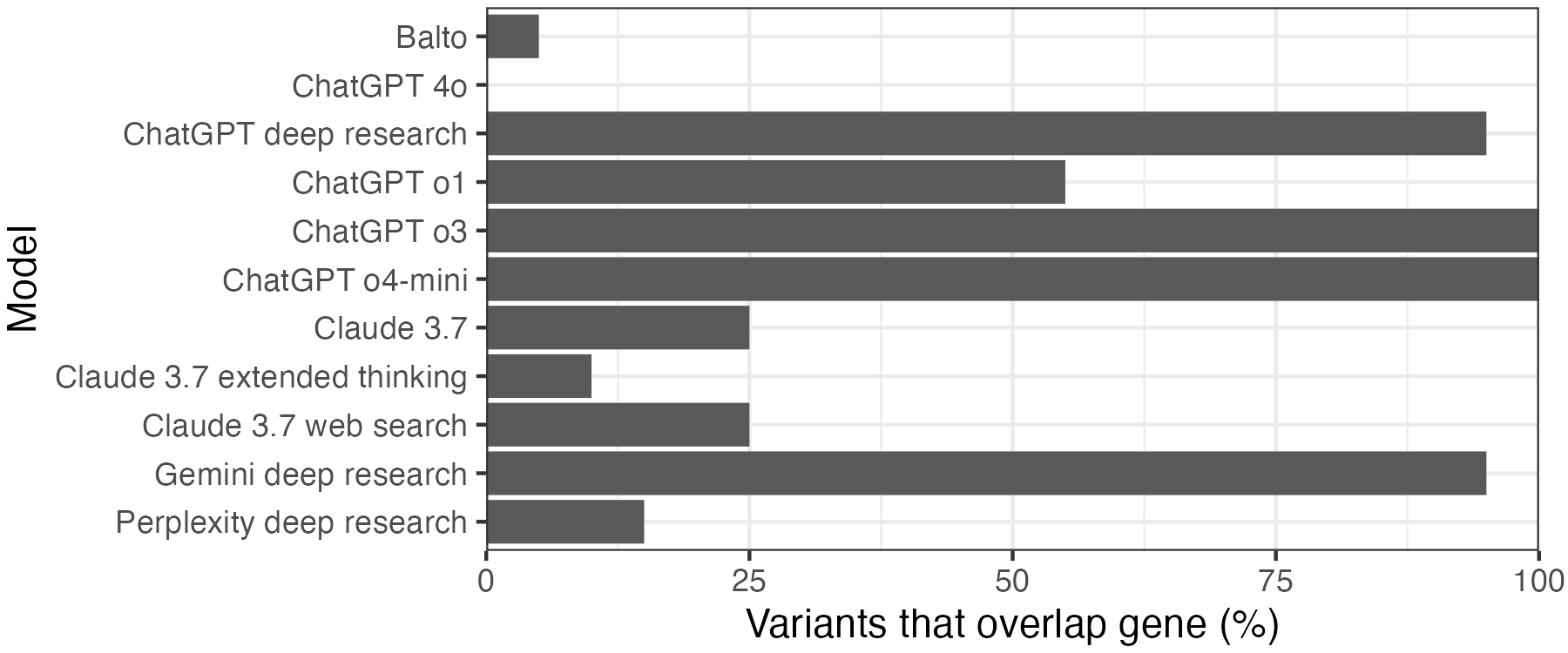
Things are slightly better if we check whether the variant at least overlaps the right gene. All of the ChatGPT o3 and o4-mini variants fall in the gene listed. Gemini and ChatGPT deep research are not far behind with 19/20. By contrast, ChatGPT 4o did not get any of the genes right, and the specialty drug discovery assistant Balto only got 1/20.
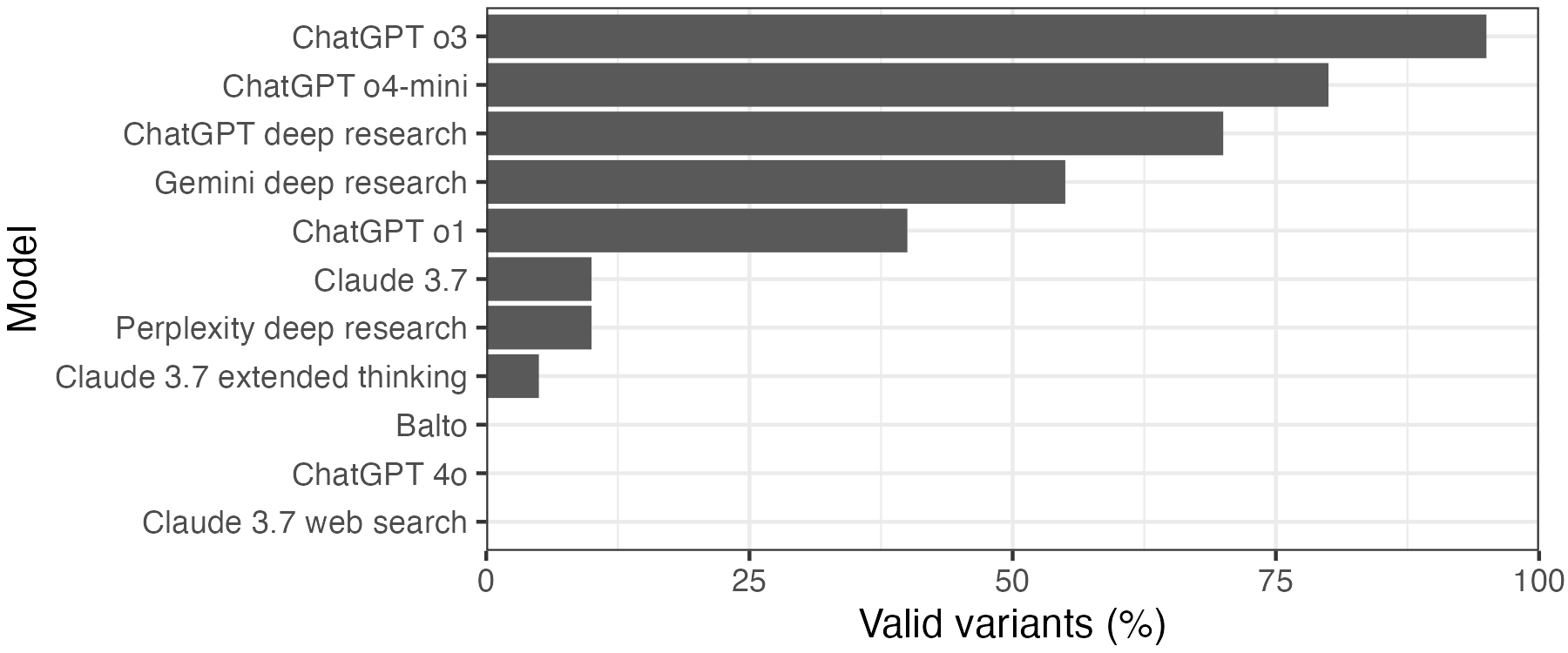
Combining these two criteria, we get a final ranking of which model yielded the most valid variants. ChatGPT o3 leads the pack, with 19/20 valid variants. 3 of 11 models did not identify any valid variants, and were excluded from further analysis.
Do the variants cause the expected splicing changes?
Next, I focused on whether the variants being reported to alter splicing are different from the ones that are reported not to alter splicing. To this end, I used SpliceAI to score all the variants and evaluated the predictive performance of the score in distinguishing splice-changing variants from other ones.
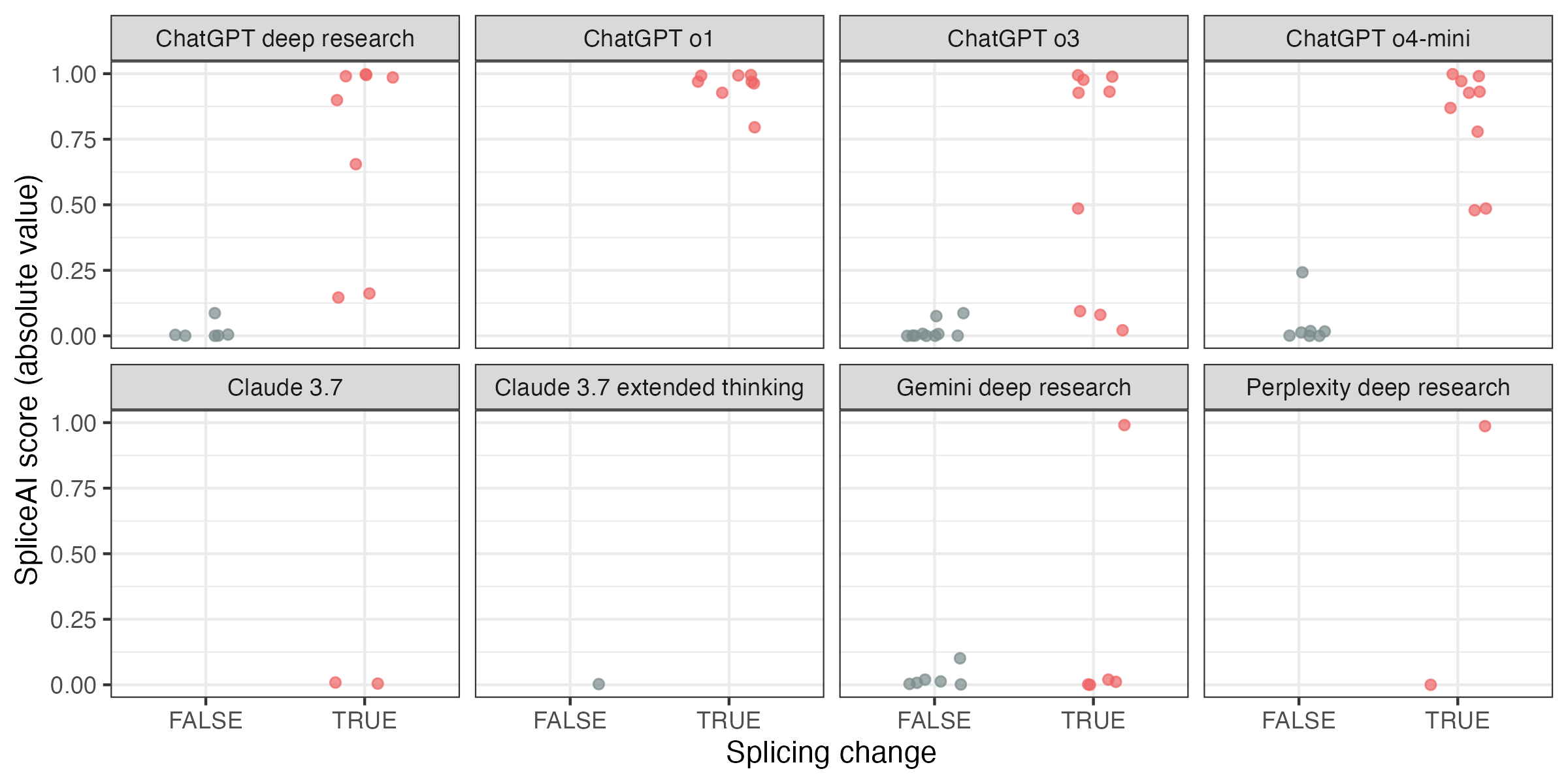
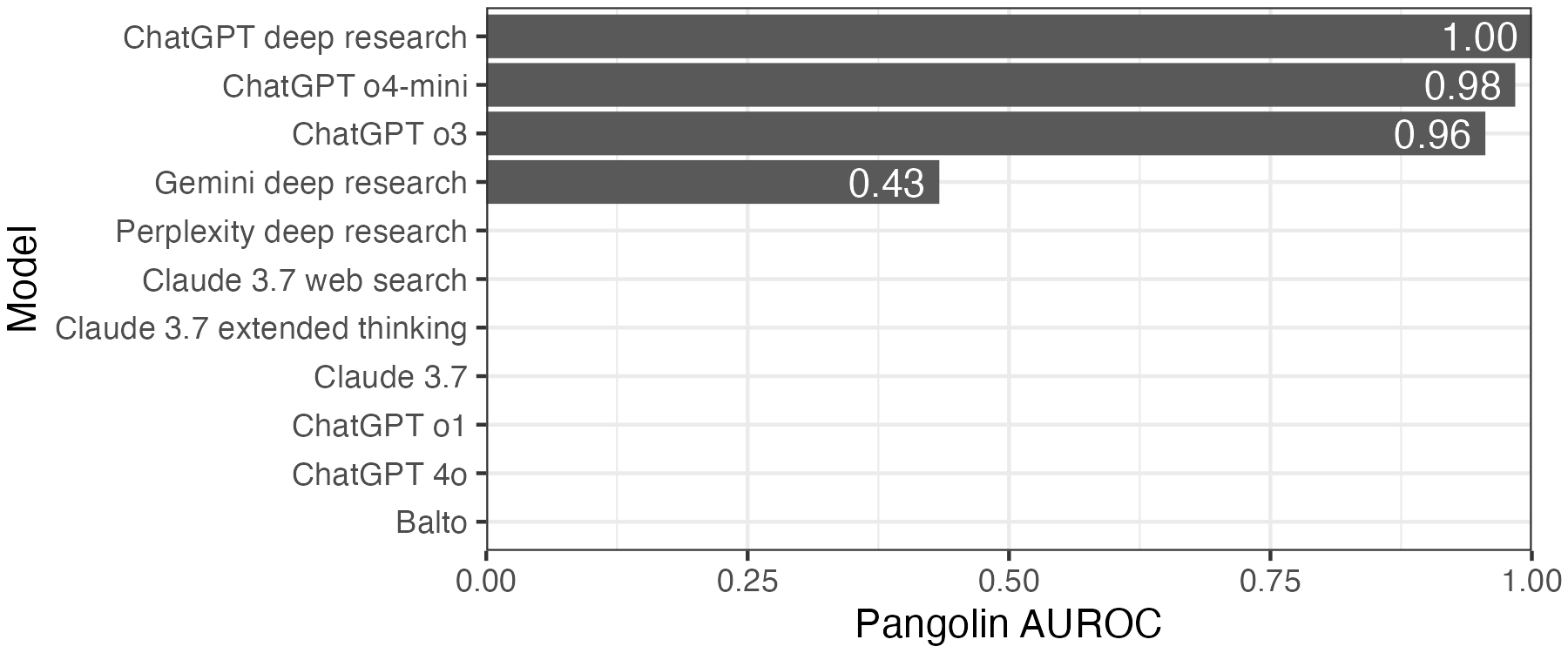
For a quantitative assessment, I calculated the AUROC. Four of the eight remaining models only produced variants of a single class, so we can only run this analysis for the other four models. Gemini performs no better than random, while the three ChatGPT models have close to perfect performance.

There’s one caveat here. SpliceAI is now routinely used for variant interpretation, introducing a possible circularity. If the scientific AI assistant has learned to extract variants based on their SpliceAI scores with no corroborating evidence, they would perform unusually well on this task. To check against this, I also scored variants with a different splicing model, Pangolin. The results are largely the same.
Conclusions
Overall, this analysis shows that models need access to literature and web searches to be able to provide accurate variant information. Within these models, the ChatGPT reasoning models o3 and o4-mini and ChatGPT deep research perform the best, while Gemini deep research is the best non-OpenAI model. The specialized scientific tools like Balto, FutureHouse Crew, and Potato do not provide any benefit, and ChatGPT Plus appears to offer the best return on investment for this use case.
Footnotes
In practice, this may not be fully accurate. If different scientific AI tend to propose variants in different classes, and the sequence-to-activity model has different performance across these classes, performance would not be constant.↩︎